| HOME | HELP | FEEDBACK | SUBSCRIPTIONS | ARCHIVE | SEARCH | TABLE OF CONTENTS |
|
| ||||||||
Volume 39 Number 20 © 2004 American Psychiatric Association p. 1
Congress Hammers FDA Over Handling of SSRIs
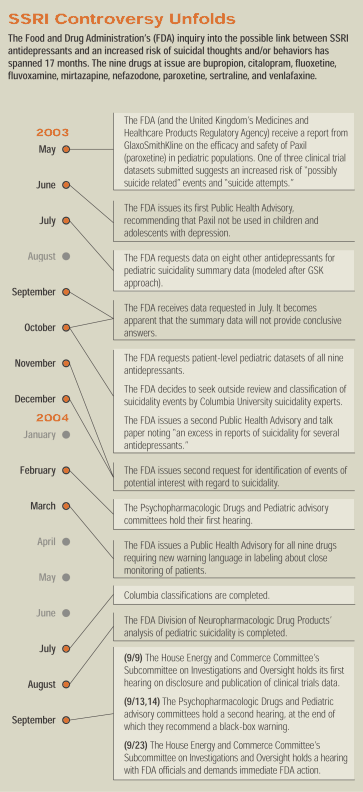
At press time, APA was waiting to hear whether the FDA would issue a black-box warning for pediatric use of SSRIs. Here is a summary of the events leading up to this strongly expected outcome. Officials of the U.S. Food and Drug Administration (FDA) first suspected an increase in suicidal thoughts and behaviors linked to antidepressant use in children and adolescents in March 1996—eight and a half years ago. Yet no apparent action was taken by the agency until a pattern seemed to emerge in the first half of 2003. During a dramatic pair of hearings before the House Energy and Commerce Committee's Subcommittee on Investigations and Oversight last month, FDA officials were repeatedly put on the defensive as to whether there existed a conspiracy to cover up the link between the drugs and harmful behaviors. Unsatisfied with answers and explanations from agency officials during the second hearing, at least two committee members, Rep. Diana DeGette (D-Colo.) and Rep. Bart Stupak (D-Mich.), threatened to introduce legislation banning the prescribing of antidepressants to anyone under age 18 "if the FDA didn't act forcefully and swiftly to protect America's children." Committee members' frustration and anger seemed to have built steadily through the first of the two hearings, which were held two weeks apart. During the first hearing, on September 9, pharmaceutical company executives were grilled about their apparent selective disclosure and publication of clinical trial findings. Only four of the 15 trials of antidepressants in pediatric patients with depression have been published—all with favorable results. Company executives fiercely defended their disclosure records, noting that the vast majority of clinical trial data not published in peer-reviewed journals—including negative data—had been presented on more than one occasion as poster presentations or symposia during major psychiatric meetings.
However, it was FDA Acting Deputy Commissioner for Operations Janet Woodcock, M.D., who took the heat from committee members. They charged that the FDA had shirked its responsibility to protect the public health by not keeping track of all the data available and ensuring disclosure. Woodcock reminded the committee members that the FDA has little statutory authority to compel disclosure or publication, fueling calls for a legislated mandatory clinical trials registry (see page 2). During the second of the two hearings, held September 23, Rep. Joe Barton (R-Tex.), chair of the Energy and Commerce Committee, emphasized Congress's frustration with the agency. "The fact that children taking antidepressants were experiencing psychiatric adverse events at greater rates than adults was known at the agency as far back as 1996 and 1997," Barton said. "I want to know what did the agency do to respond to those concerns?" Barton was referring to a previously undisclosed letter from an FDA reviewer dated March 19, 1996, in reference to Pfizer's data on the use of Zoloft (sertraline) in children and adolescents with obsessive-compulsive disorder. The FDA letter said in part, "We note that there appears to be an increased occurrence of suicidality events in pediatric patients exposed to sertraline in pediatric trials" compared with observed rates in comparable adult patients. In fact, there was a fivefold higher risk in patients taking sertraline compared with those taking placebo. The FDA medical reviewer asked Pfizer for "clarification and explanation" of the data. The subcommittee obtained the FDA's letter to Pfizer from the company, not from the agency. FDA officials acknowledged that no record of the memo was found in the agency's files. On March 28, 1996, only a week after the letter asking for clarification and explanation, the FDA approved Pfizer's application for pediatric exclusivity without any apparent resolution of the medical reviewer's questions. After the application had been approved, Pfizer responded to the agency's request. That response was retained in the FDA's files. Details also emerged about the agency's handling of a report by FDA medical reviewer Andrew Mosholder, M.D., who had analyzed the pediatric exclusivity data on antidepressants, submitted to the FDA as required under the Best Pharmaceuticals for Children Act. Mosholder's report was the first to assert openly an increased risk of suicidal thoughts and behaviors associated with antidepressant use in children and adolescents. While his analysis was instrumental in prompting an advisory committee hearing in February, Mosholder was prevented from delivering his report at that meeting. Several members of Congress, including Barton, asked numerous questions regarding the handling of Mosholder's report and why he had not been allowed to deliver it at the public advisory committee meeting. Mosholder testified that after he had completed his report, his superiors at the FDA did not agree with some of his conclusions and recommendations—he had not only identified an association, but also recommended actions that should be taken regarding strong warnings for the drugs. He testified that there was "considerable discussion" at the agency regarding the report and his interpretation of the SSRI pediatric exclusivity data. Robert Temple, M.D., the FDA's associate director for medical policy, strongly defended the agency's handling of Mosholder's report. "We were concerned about an overemphasis that could lead to a deterrent to treatment," Temple testified. "At the same time, we didn't want to ignore an important signal." Members of Congress on the oversight committee pushed Temple to explain why the agency waited so long to issue stronger warnings on SSRI use in children, when the agency had Mosholder's report since mid 2003. "We had questions that were not answered to our satisfaction," Temple said. "We were not worried about [Mosholder's] analysis of the data, but we had significant questions about the credibility of the data he had available to analyze. We decided to withhold any agency opinion on the issue till everything possible was known." The FDA then decided to seek the independent reanalysis of the data by suicidality experts at Columbia University, Temple said. "We had no way of knowing what the reanalysis would bring—whether it would strengthen or weaken [Mosholder's] conclusions. We really had no preconceived notions, no preference." Meanwhile, by late spring both House and Senate committees began investigating the FDA's handling of the issue. Members of Congress began expressing a sense of frustration over what they characterized as "a slow and obstructive response." "The FDA's lack of cooperation with the committee in obtaining relevant and responsive information in a timely fashion on a matter that involves the safety of our children leaves me wondering whether this is sheer ineptitude or something far worse," Barton noted during the first of the two hearings in September.
During the second hearing, it was revealed in documents released by the committee that on at least one occasion, documented in internal FDA e-mails between Mosholder and his superiors, Mosholder was advised to change his conclusions, deleting material on the risks. An FDA attorney then advised him to conceal the change when asked to submit documents to Congress. "I don't think it's necessary to indicate this document represents a version of the earlier one by noting that things have been omitted; that simply invites the committee to ask further questions about what was omitted," wrote Donna Katz of the FDA's Office of Chief Counsel in an e-mail to Mosholder. The Columbia analysis, which took roughly four months, was reviewed by an internal FDA Office of Drug Safety medical officer, Tarek Hammad, M.D. Hammad's conclusions mirrored those of Mosholder: there is roughly a one-and-a-half to twofold higher risk of suicidal thoughts and behaviors in children and adolescents taking certain antidepressants. "We had to proceed cautiously on such an important matter," Temple emphasized. "To get it wrong would create more problems than we could hope to solve." The Columbia analysis, he added, helped to determine that the data that Mosholder had analyzed were valid and, therefore, that his conclusions were valid. Following the second advisory committee hearings, on September 13 and 14, several members of the subcommittee pressed Temple to disclose what the FDA's final position would be (see article at right). "Stating that the drugs are associated with, linked to, or cause suicidal thoughts and behaviors in pediatric use is a given," Temple said. "What is at question is what form that warning will take. It could be a black box; it could be a bolded warning within the warnings section of the full label." Stupak shot back, "When are you finally going to decide?" Temple replied that a decision would be made "within a few days." "But if these drugs don't work well in kids, and they increase suicidality in kids, why does the FDA allow them to be prescribed to kids?" Stupak asked. "Can't the FDA ban prescribing them to children?" "We could contraindicate their use," Temple replied, "but we don't think that would be particularly helpful in protecting the public health." Temple emphasized more than once during his testimony that the agency, as well as members of the advisory committees, had been reluctant to contraindicate antidepressant use in children out of fear that children who need care would not get it. "Two-thirds of the [members of the committees] voted in favor of a black-box warning," Temple emphasized, adding that when a committee vote is split, the agency does not automatically go with the majority—it must deliberate the pros and cons of each position and determine the best course to take. "There's no question there will be a strong warning," Temple said. "We need a patient med guide, and the drugs need to be in unit dose packaging." That way, he explained, the warning language required in labeling would be assured of getting to each patient taking the medications at issue. "The only thing we are still thinking about is the box," Temple concluded. "All the rest of the advisory committee's recommendations will be implemented." A transcript of the hearing and other information will be posted
within the next few months at <http://energycommerce.house.gov/108/Hearings/09232004hearing1353/hearing.htm>.
Information on the September 13 and 14 hearing, including the
Columbia data analysis, is posted online at <www.fda.gov/ohrms/dockets/ac/cder04.html#PsychopharmacologicDrugs>.
Footnotes For developments after press time, see the Psychiatric News Web site at <pn.psychiatryonline.org>. Related articles in Psychiatric News:
|
||||||||||||||||||||||||||||||||||||||
| HOME | HELP | FEEDBACK | SUBSCRIPTIONS | ARCHIVE | SEARCH | TABLE OF CONTENTS |