| HOME | HELP | FEEDBACK | SUBSCRIPTIONS | ARCHIVE | SEARCH | TABLE OF CONTENTS |
|
| ||||||||
|
| |||||||||
| |||||||||
LIVER DISEASE |
1 Liver and Hepatobiliary Unit, Queen Elizabeth
Hospital, Birmingham, UK, and Department of Gastroenterology, Pomeranian Medical
School, Szczecin, Poland
2 Liver and Hepatobiliary Unit, Queen
Elizabeth Hospital, Birmingham, UK
3 Department of Pathology,
University of Birmingham, UK
Correspondence to:
Dr E Elias, Liver Unit, Queen
Elizabeth Hospital, 3rd floor, Nuffield House, Edgbaston, Birmingham B15 2TH,
UK;
elwyn.elias{at}uhb.nhs.uk
Accepted for publication
23 September 2002
| ABSTRACT |
|---|
Case reports: We describe the first reported case of acute cholestasis due to citalopram (selective serotonin reuptake inhibitor) occurring in a patient who also experienced obstetric cholestasis in association with each of three pregnancies; in a second patient cholestasis developed due to dothiepin (tricyclic antidepressant), and six years later due to paroxetine. In both cases liver biopsies showed features of a "pure" cholestasis with total resolution within 1–6 months after withdrawal of the causative drug. Immunostaining for the canalicular transporter, multidrug resistant protein 2 (MRP2), responsible for biliary secretion of several organic anions including bilirubin glucuronides, showed sustained expression in both biopsies as well as relocalisation with appearance of strong staining of the basolateral membrane of the hepatocyte. This finding has also not been reported previously.
Conclusions: We postulate that intracellular redistribution of MRP2 may reflect an adaptive compensatory mechanism which helps in the elimination of the drug or its cholestatic metabolites from the hepatocyte back to the sinusoidal space and subsequent excretion in urine. Changes seen in these two patients differ from findings previously reported in rats where downregulation of mrp2 occurs in response to experimentally induced cholestasis. We speculate that the rat is more advanced than humans in its ability to downregulate canalicular transporter expression as protection against progressive intrahepatic cholestasis.
Keywords: antidepressants; cholestasis; multidrug resistant protein; MRP2
Abbreviations: MRP2, multidrug resistant protein 2; SSRI, selective serotonin reuptake inhibitor; LFTs, liver function tests; ALP, alkaline phosphatase; AST, aspartate transaminase; INR, international normalised ratio; SBA, serum bile acids; TBS, Tris buffered saline; OC, obstetric cholestasis
Antidepressive drugs may occasionally cause impairment of liver function. Both tricyclic antidepressant and monoamine oxidase inhibitors have been reported to induce prolonged1 or even fatal jaundice.2,3 Selective serotonin reuptake inhibitors (SSRI), such as paroxetine, have been shown to induce severe acute or chronic hepatitis.4,5 The mechanisms by which antidepressive drugs induce cholestatic changes are not known. Recent cloning of proteins involved in the secretion of bile and its constituents has helped to provide a better understanding of the processes involved in the pathogenesis of some cholestatic disorders.6 However, data on expression of these transporters in drug induced cholestasis do not exist.6
In this paper, we describe two cases of antidepressant induced cholestasis. The first patient developed acute cholestasis during treatment with citalopram, an SSRI, which is the first report of such a cholestatic reaction. The second patient developed features of cholestasis after dothiepin (tricyclic antidepressant) and six years later he presented with similar symptoms associated with treatment with paroxetine. Neither the occurrence of two episodes of cholestasis following two different antidepressants in one patient nor acute cholestasis caused by paroxetine have been reported previously. We also looked (for the first time in drug induced cholestasis) at the hepatocellular distribution of multidrug resistant protein 2 (MRP2), one of the key canalicular proteins responsible for the transport of several organic anions, including bilirubin glucuronides, from the hepatocyte to bile.
| PATIENTS |
|---|
|
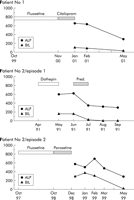
In October 1997 he received fluoxetine and remained on it for 12
months. It did not affect his depression and therefore his local
general practitioner switched him to paroxetine at a dose of 20 mg
daily. Two months later he developed symptoms of cholestasis and
presented with jaundice and pruritus. His LFTs showed bilirubin 260
µmol/l, ALP 544 U/l, AST 36 U/l, SBA 456 (normal <15), albumin 43
g/l, and INR 1.0. As in the first episode of cholestasis,
autoantibody and viral screening were negative and abdominal
ultrasound was normal. Paroxetine was stopped and his symptoms
resolved within a couple of months. His LFTs in May 1999 were
as follows: bilirubin 7 µmol/l; ALP 255 U/l; AST 35 U/l; SBA
15; albumin 43; and INR 1.0. He was recently seen in our clinic in
May 2001 (fig 1C![]() ). His
LFTs remain entirely normal and with respect to his liver, he is
asymptomatic.
). His
LFTs remain entirely normal and with respect to his liver, he is
asymptomatic.
| IMMUNOHISTOCHEMISTRY |
|---|
Results
Histologically normal liver tissue
obtained from donor liver used for transplantation was used as a
normal control. This showed diffuse canalicular immunostaining for
MRP2 with no obvious zonal variation in staining intensity (fig 2A![]() ).
Immunostaining was also present in the epithelium of bile ducts and
ductules. This was mainly luminal with focal basolateral staining
being evident.
).
Immunostaining was also present in the epithelium of bile ducts and
ductules. This was mainly luminal with focal basolateral staining
being evident.
|
| DISCUSSION |
|---|
The striking zonal distribution of canalicular plugging supports
the theory that in these cholestatic drug reactions biliary
bile acids are insufficient for solubilisation of biliary solutes
which form intracanalicular precipitates in lobular zones 3 and
2 but sufficient to prevent canalicular precipitates accumulating in
zone1.10
The severe degree of bile plugging seen in both our patients is not
unusual in several human cholestatic conditions, including those
caused by sepsis, drugs, and haemolysis, but not classically seen in
animal models of cholestasis. This suggests species differences in
the control and regulation of biliary transporter proteins. In the
rat, downregulation of mrp2, the transporter responsible for
canalicular secretion of bilirubin, has been shown to be an early
response to a variety of cholestatic insults in models using
endotoxin, oestradiol, and bile duct ligated rats.11
This downregulation would protect the rodent liver against the kind
of biliary plugging seen in our patients. Regulation of mrp2 and ntcp
expression in the rat occurs via RXR/RAR and HNF-1, downregulation
occurring in response to bile acid ligands as well as in response to
cytokines, and is associated with nuclear factor  B stimulated
upregulation of Mdr1b. Our immunohistological observations provide no
evidence in support of MRP2 downregulation in our severely
cholestatic patients. The high serum bile acid level of >400 is of
an order we see in patients with complete deficiency of expression of
BSEP and suggests that biliary plugging has resulted from
maintained levels of bilirubin and other organic anion secretion
despite poor biliary bile acid output. In turn, it suggests that
human MRP2 expression is not as susceptible to downregulation as
it is in the rat, which may therefore be perceived as more
advanced in its hepatoprotective repertoire of gene expression
which confers protection against xenobiotics.
B stimulated
upregulation of Mdr1b. Our immunohistological observations provide no
evidence in support of MRP2 downregulation in our severely
cholestatic patients. The high serum bile acid level of >400 is of
an order we see in patients with complete deficiency of expression of
BSEP and suggests that biliary plugging has resulted from
maintained levels of bilirubin and other organic anion secretion
despite poor biliary bile acid output. In turn, it suggests that
human MRP2 expression is not as susceptible to downregulation as
it is in the rat, which may therefore be perceived as more
advanced in its hepatoprotective repertoire of gene expression
which confers protection against xenobiotics.
Our immunostaining study performed on both biopsies showed significant redistribution of one of the key canalicular transporters, multidrug resistant protein 2 (MRP2). MRP2 is a recently cloned protein responsible for the canalicular transport of several organic anions, including bilirubin. We have tested a wide range of antibodies against hepatocellular transporters but it was only MRP2 antibody which worked in paraffin embedded sections. Despite the fact that in both biopsies analysed cholestasis was induced by two different agents, MRP2 stainings showed striking similarities, especially in their strong basolateral membrane reaction, not seen in normal liver. Redistribution of MRP2 into the basolateral membrane has been observed in vitro in HepG2 cells.12 Under physiological conditions, transport of MRP2 from the Golgi apparatus to the canaliculus occurs via a sinusoidal membrane, as demonstrated by Boyer and Soroka.13 One may assume that membranous staining observed by us may be explained by cholestasis induced disturbance of the polarity of hepatocytes. We have recently shown however that in another cholestatic condition, primary biliary cirrhosis, MRP2 maintained its characteristic and specifically canalicular distribution without any detectable staining of the basolateral membrane of the hepatocyte14 which would be indicative of lost hepatocellular polarity. It seems therefore more likely that basolateral membrane expression of MRP2 reflects an adaptive compensatory mechanism which helps in elimination of the drug or its cholestatic metabolites from the hepatocyte back to the sinusoidal space, and then the subsequent excretion of the drug (metabolite) in urine. Our finding that MDR3, a phospholipid flippase, is uncharacteristically redistributed to the basolateral membrane of the hepatocyte in the periphery of cirrhotic nodules in advanced primary biliary cirrhosis, in association with upregulation of MRP3 which is able to excrete bile acids into the blood, was interpreted by us as an adaptive response of the cholestatic hepatocyte facilitating extrusion of bile acids and "biliary" phospholipid into plasma.14 We can postulate that the parallel uncharacteristic basolateral expression of MRP2 in our patients is a response of the hepatocyte to an extreme degree of canalicular obstruction. Whether the redistribution occurs prior to insertion of the transporter into the canalicular membrane or subsequently by lateral diffusion from the canalicular domain via tight junctions rendered incompetent as a consequence of the drug toxicity per se, or secondary to plugging, remains to be determined.
It is likely that antidepressant induced cholestasis may occur in predisposed individuals. The observation that patient No 1 developed OC in addition to citalopram jaundice and patient No 2 developed cholestatic jaundice in response to two drugs of different classes strongly supports the hypothesis that they represent genetically predisposed individuals who are susceptible to cholestatic jaundice which can be precipitated by a variety of stimuli. The predisposed liver thus responds with a limited repertoire to diverse insults. We were not able to identify predisposing factors in patient No 2. Patient No 1 however presented a prior history of OC. In fact, she became pregnant soon after recovery from her citalopram induced cholestasis and again developed features of OC. OC can be associated with malfunction of other canalicular transporters, or a mutation causing disturbed trafficking of MDR3 to the canalicular membrane, as recently shown by us and others.15,16 Whether a molecular defect(s) leading to OC predisposes to SSRI induced cholestasis remains to be elucidated.
| REFERENCES |
|---|
| |||||||||
| HOME | HELP | FEEDBACK | SUBSCRIPTIONS | ARCHIVE | SEARCH | TABLE OF CONTENTS |
 )
)