 |
PDR® entry for
STRATTERA™
(Lilly)
(atomoxetine HCl)
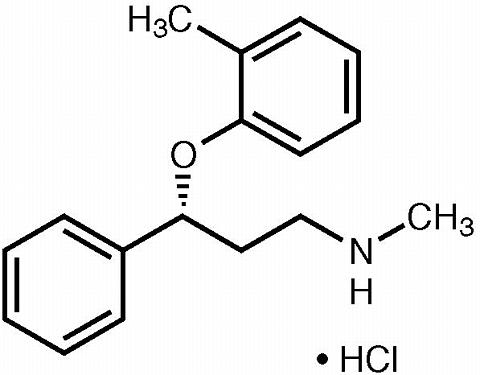
STRATTERA™ (atomoxetine HCl) is a selective norepinephrine reuptake inhibitor. Atomoxetine HCl is the R (-) isomer as determined by x-ray diffraction. The chemical designation is (-)- N -methyl-3-phenyl-3-( o -tolyloxy)-propylamine hydrochloride. The molecular formula is C 17 H 21 NO•HCl, which corresponds to a molecular weight of 291.82. The chemical structure is:
|
Atomoxetine HCl is a white to practically white solid, which has a solubility of 27.8 mg/mL in water.
STRATTERA capsules are intended for oral administration only.
Each capsule contains atomoxetine HCl equivalent to 10, 18, 25, 40, or 60 mg of atomoxetine. The capsules also contain pregelatinized starch and dimethicone. The capsule shells contain gelatin, sodium lauryl sulfate, and other inactive ingredients. The capsule shells also contain one or more of the following: FD&C Blue No. 2, synthetic yellow iron oxide, titanium dioxide. The capsules are imprinted with edible black ink.
The precise mechanism by which atomoxetine produces its therapeutic effects in Attention-Deficit/Hyperactivity Disorder (ADHD) is unknown, but is thought to be related to selective inhibition of the pre-synaptic norepinephrine transporter, as determined in ex vivo uptake and neurotransmitter depletion studies.
Atomoxetine is well-absorbed after oral administration and is minimally affected by food. It is eliminated primarily by oxidative metabolism through the cytochrome P450 2D6 (CYP2D6) enzymatic pathway and subsequent glucuronidation. Atomoxetine has a half-life of about 5 hours. A fraction of the population (about 7% of Caucasians and 2% of African Americans) are poor metabolizers (PMs) of CYP2D6 metabolized drugs. These individuals have reduced activity in this pathway resulting in 10-fold higher AUCs, 5-fold higher peak plasma concentrations, and slower elimination (plasma half-life of about 24 hours) of atomoxetine compared with people with normal activity [extensive metabolizers (EMs)]. Drugs that inhibit CYP2D6, such as fluoxetine, paroxetine, and quinidine, cause similar increases in exposure.
The pharmacokinetics of atomoxetine have been evaluated in more than 400 children and adolescents in selected clinical trials, primarily using population pharmacokinetic studies. Single-dose and steady-state individual pharmacokinetic data were also obtained in children, adolescents, and adults. When doses were normalized to a mg/kg basis, similar half-life, C max , and AUC values were observed in children, adolescents, and adults. Clearance and volume of distribution after adjustment for body weight were also similar.
Absorption and Distribution --Atomoxetine is rapidly absorbed after oral administration, with absolute bioavailability of about 63% in EMs and 94% in PMs. Maximal plasma concentrations (C max ) are reached approximately 1 to 2 hours after dosing.
STRATTERA can be administered with or without food. Administration of STRATTERA with a standard high-fat meal in adults did not affect the extent of oral absorption of atomoxetine (AUC), but did decrease the rate of absorption, resulting in a 37% lower C max , and delayed T max by 3 hours. In clinical trials with children and adolescents, administration of STRATTERA with food resulted in a 9% lower C max .
The steady-state volume of distribution after intravenous administration is 0.85 L/kg indicating that atomoxetine distributes primarily into total body water. Volume of distribution is similar across the patient weight range after normalizing for body weight.
At therapeutic concentrations, 98% of atomoxetine in plasma is bound to protein, primarily albumin.
Metabolism and Elimination --Atomoxetine is metabolized primarily through the CYP2D6 enzymatic pathway. People with reduced activity in this pathway (PMs) have higher plasma concentrations of atomoxetine compared with people with normal activity (EMs). For PMs, AUC of atomoxetine is approximately 10-fold and C ss,max is about 5-fold greater than EMs. Laboratory tests are available to identify CYP2D6 PMs. Coadministration of STRATTERA with potent inhibitors of CYP2D6, such as fluoxetine, paroxetine, or quinidine, results in a substantial increase in atomoxetine plasma exposure, and dosing adjustment may be necessary ( see Drug-Drug Interactions ). Atomoxetine did not inhibit or induce the CYP2D6 pathway.
The major oxidative metabolite formed, regardless of CYP2D6 status, is 4-hydroxyatomoxetine, which is glucuronidated. 4-Hydroxyatomoxetine is equipotent to atomoxetine as an inhibitor of the norepinephrine transporter but circulates in plasma at much lower concentrations (1% of atomoxetine concentration in EMs and 0.1% of atomoxetine concentration in PMs). 4-Hydroxyatomoxetine is primarily formed by CYP2D6, but in PMs, 4-hydroxyatomoxetine is formed at a slower rate by several other cytochrome P450 enzymes. N-Desmethylatomoxetine is formed by CYP2C19 and other cytochrome P450 enzymes, but has substantially less pharmacological activity compared with atomoxetine and circulates in plasma at lower concentrations (5% of atomoxetine concentration in EMs and 45% of atomoxetine concentration in PMs).
Mean apparent plasma clearance of atomoxetine after oral administration in adult EMs is 0.35 L/hr/kg and the mean half-life is 5.2 hours. Following oral administration of atomoxetine to PMs, mean apparent plasma clearance is 0.03 L/hr/kg and mean half-life is 21.6 hours. For PMs, AUC of atomoxetine is approximately 10-fold and C ss,max is about 5-fold greater than EMs. The elimination half-life of 4-hydroxyatomoxetine is similar to that of N-desmethylatomoxetine (6 to 8 hours) in EM subjects, while the half-life of N-desmethylatomoxetine is much longer in PM subjects (34 to 40 hours).
Atomoxetine is excreted primarily as 4-hydroxyatomoxetine- O -glucuronide, mainly in the urine (greater than 80% of the dose) and to a lesser extent in the feces (less than 17% of the dose). Only a small fraction of the STRATTERA dose is excreted as unchanged atomoxetine (less than 3% of the dose), indicating extensive biotransformation.
Hepatic insufficiency --Atomoxetine exposure (AUC) is increased, compared with normal subjects, in EM subjects with moderate (Child-Pugh Class B) (2-fold increase) and severe (Child-Pugh Class C) (4-fold increase) hepatic insufficiency. Dosage adjustment is recommended for patients with moderate or severe hepatic insufficiency ( see DOSAGE AND ADMINISTRATION ).
Renal insufficiency --EM subjects with end stage renal disease had higher systemic exposure to atomoxetine than healthy subjects (about a 65% increase), but there was no difference when exposure was corrected for mg/kg dose. STRATTERA can therefore be administered to ADHD patients with end stage renal disease or lesser degrees of renal insufficiency using the normal dosing regimen.
Geriatric --The pharmacokinetics of atomoxetine have not been evaluated in the geriatric population.
Pediatric --The pharmacokinetics of atomoxetine in children and adolescents are similar to those in adults. The pharmacokinetics of atomoxetine have not been evaluated in children under 6 years of age.
Gender --Gender did not influence atomoxetine disposition.
Ethnic origin --Ethnic origin did not influence atomoxetine disposition (except that PMs are more common in Caucasians).
CYP2D6 activity and atomoxetine plasma concentration --Atomoxetine is primarily metabolized by the CYP2D6 pathway to 4-hydroxyatomoxetine. In EMs, inhibitors of CYP2D6 increase atomoxetine steady-state plasma concentrations to exposures similar to those observed in PMs. Dosage adjustment of STRATTERA in EMs may be necessary when coadministered with CYP2D6 inhibitors, e.g., paroxetine, fluoxetine, and quinidine ( see Drug Interactions under PRECAUTIONS ). In vitro studies suggest that coadministration of cytochrome P450 inhibitors to PMs will not increase the plasma concentrations of atomoxetine.
Effect of atomoxetine on P450 enzymes --Atomoxetine did not cause clinically important inhibition or induction of cytochrome P450 enzymes, including CYP1A2, CYP3A, CYP2D6, and CYP2C9.
Albuterol --Albuterol (600 mcg iv over 2 hours) induced increases in heart rate and blood pressure. These effects were potentiated by atomoxetine (60 mg BID for 5 days) and were most marked after the initial coadministration of albuterol and atomoxetine ( see Drug-Drug Interactions under PRECAUTIONS ).
Alcohol --Consumption of ethanol with STRATTERA did not change the intoxicating effects of ethanol.
Desipramine --Coadministration of STRATTERA (40 or 60 mg BID for 13 days) with desipramine, a model compound for CYP2D6 metabolized drugs (single dose of 50 mg), did not alter the pharmacokinetics of desipramine. No dose adjustment is recommended for drugs metabolized by CYP2D6.
Methylphenidate --Coadministration of methylphenidate with STRATTERA did not increase cardiovascular effects beyond those seen with methylphenidate alone.
Midazolam --Coadministration of STRATTERA (60 mg BID for 12 days) with midazolam, a model compound for CYP3A4 metabolized drugs, (single dose of 5 mg), resulted in 15% increase in AUC of midazolam. No dose adjustment is recommended for drugs metabolized by CYP3A.
Drugs highly bound to plasma protein --In vitro drug-displacement studies were conducted with atomoxetine and other highly-bound drugs at therapeutic concentrations. Atomoxetine did not affect the binding of warfarin, acetylsalicylic acid, phenytoin, or diazepam to human albumin. Similarly, these compounds did not affect the binding of atomoxetine to human albumin.
Drugs that affect gastric pH --Drugs that elevate gastric pH (magnesium hydroxide/aluminum hydroxide, omeprazole) had no effect on STRATTERA bioavailability.
The effectiveness of STRATTERA in the treatment of ADHD was established in 6 randomized, double-blind, placebo-controlled studies in children, adolescents, and adults who met Diagnostic and Statistical Manual 4 th edition (DSM-IV) criteria for ADHD ( see INDICATIONS AND USAGE ).
The effectiveness of STRATTERA in the treatment of ADHD was established in 4 randomized, double-blind, placebo-controlled studies of pediatric patients (ages 6 to 18). Approximately one-third of the patients met DSM-IV criteria for inattentive subtype and two-thirds met criteria for both inattentive and hyperactive/impulsive subtypes ( see INDICATIONS AND USAGE ).
Signs and symptoms of ADHD were evaluated by a comparison of mean change from baseline to endpoint for STRATTERA- and placebo-treated patients using an intent-to-treat analysis of the primary outcome measure, the investigator administered and scored ADHD Rating Scale-IV-Parent Version (ADHDRS) total score including hyperactive/impulsive and inattentive subscales. Each item on the ADHDRS maps directly to one symptom criterion for ADHD in the DSM-IV.
In Study 1, an 8-week randomized, double-blind, placebo-controlled, dose-response, acute treatment study of children and adolescents aged 8 to 18 (N=297), patients received either a fixed dose of STRATTERA (0.5, 1.2, or 1.8 mg/kg/day) or placebo. STRATTERA was administered as a divided dose in the early morning and late afternoon/early evening. At the 2 higher doses, improvements in ADHD symptoms were statistically significantly superior in STRATTERA-treated patients compared with placebo-treated patients as measured on the ADHDRS scale. The 1.8-mg/kg/day STRATTERA dose did not provide any additional benefit over that observed with the 1.2-mg/kg/day dose. The 0.5-mg/kg/day STRATTERA dose was not superior to placebo.
In Study 2, a 6-week randomized, double-blind, placebo-controlled, acute treatment study of children and adolescents aged 6 to 16 (N=171), patients received either STRATTERA or placebo. STRATTERA was administered as a single dose in the early morning and titrated on a weight-adjusted basis according to clinical response, up to a maximum dose of 1.5 mg/kg/day. The mean final dose of STRATTERA was approximately 1.3 mg/kg/day. ADHD symptoms were statistically significantly improved on STRATTERA compared with placebo, as measured on the ADHDRS scale. This study shows that STRATTERA is effective when administered once daily in the morning.
In 2 identical, 9-week, acute, randomized, double-blind, placebo-controlled studies of children aged 7 to 13 (Study 3, N=147; Study 4, N=144), STRATTERA and methylphenidate were compared with placebo. STRATTERA was administered as a divided dose in the early morning and late afternoon (after school) and titrated on a weight-adjusted basis according to clinical response. The maximum recommended STRATTERA dose was 2.0 mg/kg/day. The mean final dose of STRATTERA for both studies was approximately 1.6 mg/kg/day. In both studies, ADHD symptoms statistically significantly improved more on STRATTERA than on placebo, as measured on the ADHDRS scale.
Examination of population subsets based on gender and age (<12 and 12 to 17) did not reveal any differential responsiveness on the basis of these subgroupings. There was not sufficient exposure of ethnic groups other than Caucasian to allow exploration of differences in these subgroups.
The effectiveness of STRATTERA in the treatment of ADHD was established in 2 randomized, double-blind, placebo-controlled clinical studies of adult patients, age 18 and older, who met DSM-IV criteria for ADHD.
Signs and symptoms of ADHD were evaluated using the investigator-administered Conners Adult ADHD Rating Scale Screening Version (CAARS), a 30-item scale. The primary effectiveness measure was the 18-item Total ADHD Symptom score (the sum of the inattentive and hyperactivity/impulsivity subscales from the CAARS) evaluated by a comparison of mean change from baseline to endpoint using an intent-to-treat analysis.
In 2 identical, 10-week, randomized, double-blind, placebo-controlled acute treatment studies (Study 5, N=280; Study 6, N=256), patients received either STRATTERA or placebo. STRATTERA was administered as a divided dose in the early morning and late afternoon/early evening and titrated according to clinical response in a range of 60 to 120 mg/day. The mean final dose of STRATTERA for both studies was approximately 95 mg/day. In both studies, ADHD symptoms were statistically significantly improved on STRATTERA, as measured on the ADHD Symptom score from the CAARS scale.
Examination of population subsets based on gender and age (<42 and >/=42) did not reveal any differential responsiveness on the basis of these subgroupings. There was not sufficient exposure of ethnic groups other than Caucasian to allow exploration of differences in these subgroups.
STRATTERA is indicated for the treatment of Attention-Deficit/Hyperactivity Disorder (ADHD).
The effectiveness of STRATTERA in the treatment of ADHD was established in 2 placebo-controlled trials in children, 2 placebo-controlled trials in children and adolescents, and 2 placebo-controlled trials in adults who met DSM-IV criteria for ADHD ( see CLINICAL STUDIES ).
A diagnosis of ADHD (DSM-IV) implies the presence of hyperactive-impulsive or inattentive symptoms that cause impairment and that were present before age 7 years. The symptoms must be persistent, must be more severe than is typically observed in individuals at a comparable level of development, must cause clinically significant impairment, e.g., in social, academic, or occupational functioning, and must be present in 2 or more settings, e.g., school (or work) and at home. The symptoms must not be better accounted for by another mental disorder. For the Inattentive Type, at least 6 of the following symptoms must have persisted for at least 6 months: lack of attention to details/careless mistakes, lack of sustained attention, poor listener, failure to follow through on tasks, poor organization, avoids tasks requiring sustained mental effort, loses things, easily distracted, forgetful. For the Hyperactive-Impulsive Type, at least 6 of the following symptoms must have persisted for at least 6 months: fidgeting/squirming, leaving seat, inappropriate running/climbing, difficulty with quiet activities, "on the go," excessive talking, blurting answers, can't wait turn, intrusive. For a Combined Type diagnosis, both inattentive and hyperactive-impulsive criteria must be met.
The specific etiology of ADHD is unknown, and there is no single diagnostic test. Adequate diagnosis requires the use not only of medical but also of special psychological, educational, and social resources. Learning may or may not be impaired. The diagnosis must be based upon a complete history and evaluation of the patient and not solely on the presence of the required number of DSM-IV characteristics.
STRATTERA is indicated as an integral part of a total treatment program for ADHD that may include other measures (psychological, educational, social) for patients with this syndrome. Drug treatment may not be indicated for all patients with this syndrome. Drug treatment is not intended for use in the patient who exhibits symptoms secondary to environmental factors and/or other primary psychiatric disorders, including psychosis. Appropriate educational placement is essential in children and adolescents with this diagnosis and psychosocial intervention is often helpful. When remedial measures alone are insufficient, the decision to prescribe drug treatment medication will depend upon the physician's assessment for the chronicity and severity of the patient's symptoms.
The effectiveness of STRATTERA for long-term use, ie, for more than 9 weeks in child and adolescent patients and 10 weeks in adult patients, has not been systematically evaluated in controlled trials. Therefore, the physician who elects to use STRATTERA for extended periods should periodically reevaluate the long-term usefulness of the drug for the individual patient ( see DOSAGE AND ADMINISTRATION ).
STRATTERA is contraindicated in patients known to be hypersensitive to atomoxetine or other constituents of the product ( see WARNINGS ).
STRATTERA should not be taken with an MAOI, or within 2 weeks after discontinuing an MAOI. Treatment with an MAOI should not be initiated within 2 weeks after discontinuing STRATTERA. With other drugs that affect brain monoamine concentrations, there have been reports of serious, sometimes fatal, reactions (including hyperthermia, rigidity, myoclonus, autonomic instability with possible rapid fluctuations of vital signs, and mental status changes that include extreme agitation progressing to delirium and coma) when taken in combination with an MAOI. Some cases presented with features resembling neuroleptic malignant syndrome. Such reactions may occur when these drugs are given concurrently or in close proximity.
In clinical trials, STRATTERA use was associated with an increased risk of mydriasis and therefore its use is not recommended in patients with narrow angle glaucoma.
Although uncommon, allergic reactions, including angioneurotic edema, urticaria, and rash, have been reported in patients taking STRATTERA.
Growth should be monitored during treatment with STRATTERA. During acute treatment studies (up to 9 weeks), STRATTERA-treated patients lost an average of 0.4 kg, while placebo patients gained an average of 1.5 kg. In a controlled trial that randomized patients to placebo or 1 of 3 atomoxetine doses, 1.3%, 7.1%, 19.3%, and 29.1% of patients lost at least 3.5% of their body weight in the placebo, 0.5, 1.2, and 1.8 mg/kg/day STRATTERA dose groups, respectively. During acute treatment studies, STRATTERA-treated patients grew an average of 0.9 cm, while placebo-treated patients grew an average of 1.1 cm. There are no long-term, placebo-controlled data to evaluate the effect of STRATTERA on growth. Weight and height were assessed during open-label studies of 12 and 18 months, and mean rates of growth were compared to normal growth curves. Patients treated with STRATTERA for at least 18 months gained an average of 6.5 kg while mean weight percentile decreased slightly from 68 to 60. For this same group of patients, the average gain in height was 9.3 cm with a slight decrease in mean height percentile from 54 to 50. Among patients treated for at least 6 months, mean weight gain was lower for poor metabolizer (PM) patients compared with extensive metabolizer (EM) patients (+0.7 kg compared with +3.0 kg), while mean growth for PM patients was 4.3 cm and mean growth for EM patients was 4.4 cm. Whether final adult height or weight is affected by treatment with STRATTERA is unknown. Patients requiring long-term therapy should be monitored, and consideration should be given to interrupting therapy in patients who are not growing or gaining weight satisfactorily.
Effects on blood pressure and heart rate --STRATTERA should be used with caution in patients with hypertension, tachycardia, or cardiovascular or cerebrovascular disease because it can increase blood pressure and heart rate. Pulse and blood pressure should be measured at baseline, following STRATTERA dose increases, and periodically while on therapy.
In pediatric placebo-controlled trials, STRATTERA-treated subjects experienced a mean increase in heart rate of about 6 beats/minute compared with placebo subjects. At the final study visit before drug discontinuation, 3.6% (12/335) of STRATTERA-treated subjects had heart rate increases of at least 25 beats/minute and a heart rate of at least 110 beats/minute, compared with 0.5% (1/204) of placebo subjects. No pediatric subject had a heart rate increase of at least 25 beats/minute and a heart rate of at least 110 beats/minute on more than one occasion. Tachycardia was identified as an adverse event for 1.5% (5/340) of these pediatric subjects compared with 0.5% (1/207) of placebo subjects. The mean heart rate increase in extensive metabolizer (EM) patients was 6.7 beats/minute, and in poor metabolizer (PM) patients 10.4 beats/minute.
STRATTERA-treated pediatric subjects experienced mean increases of about 1.5 mm Hg in systolic and diastolic blood pressures compared with placebo. At the final study visit before drug discontinuation, 6.8% (22/324) of STRATTERA-treated pediatric subjects had high systolic blood pressure measurements compared with 3.0% (6/197) of placebo subjects. High systolic blood pressures were measured on 2 or more occasions in 8.6% (28/324) of STRATTERA-treated subjects and 3.6% (7/197) of placebo subjects. At the final study visit before drug discontinuation, 2.8% (9/326) of STRATTERA-treated pediatric subjects had high diastolic blood pressure measurements compared with 0.5% (1/200) of placebo subjects. High diastolic blood pressures were measured on 2 or more occasions in 5.2% (17/326) of STRATTERA-treated subjects and 1.5% (3/200) placebo subjects. (High systolic and diastolic blood pressure measurements were defined as those exceeding the 95 th percentile, stratified by age, gender, and height percentile - National High Blood Pressure Education Working Group on Hypertension Control in Children and Adolescents.)
In adult placebo-controlled trials, STRATTERA-treated subjects experienced a mean increase in heart rate of 5 beats/minute compared with placebo subjects. Tachycardia was identified as an adverse event for 3% (8/269) of these adult atomoxetine subjects compared with 0.8% (2/263) of placebo subjects.
STRATTERA-treated adult subjects experienced mean increases in systolic (about 3 mm Hg) and diastolic (about 1 mm Hg) blood pressures compared with placebo. At the final study visit before drug discontinuation, 1.9% (5/258) of STRATTERA-treated adult subjects had systolic blood pressure measurements >/=150 mm Hg compared with 1.2% (3/256) of placebo subjects. At the final study visit before drug discontinuation, 0.8% (2/257) of STRATTERA-treated adult subjects had diastolic blood pressure measurements >/=100 mm Hg compared with 0.4% (1/257) of placebo subjects. No adult subject had a high systolic or diastolic blood pressure detected on more than one occasion.
Orthostatic hypotension has been reported in subjects taking STRATTERA. In short-term child- and adolescent-controlled trials, 1.8% (6/340) of STRATTERA-treated subjects experienced symptoms of postural hypotension compared with 0.5% (1/207) of placebo-treated subjects. STRATTERA should be used with caution in any condition that may predispose patients to hypotension.
Effects on urine outflow from the bladder --In adult ADHD controlled trials, the rates of urinary retention (3%, 7/269) and urinary hesitation (3%, 7/269) were increased among atomoxetine subjects compared with placebo subjects (0%, 0/263). Two adult atomoxetine subjects and no placebo subjects discontinued from controlled clinical trials because of urinary retention. A complaint of urinary retention or urinary hesitancy should be considered potentially related to atomoxetine.
Patients should read Information for Patients before starting therapy with STRATTERA and when the prescription is renewed.
Patients should consult a physician if they are taking or plan to take any prescription or over-the-counter medicines, dietary supplements, or herbal remedies.
Patients should consult a physician if they are nursing, pregnant, or thinking of becoming pregnant while taking STRATTERA.
Patients may take STRATTERA with or without food.
If patients miss a dose, they should take it as soon as possible, but should not take more than the prescribed total daily amount of STRATTERA in any 24-hour period.
Patients should use caution when driving a car or operating hazardous machinery until they are reasonably certain that their performance is not affected by atomoxetine.
Routine laboratory tests are not required.
CYP2D6 metabolism --Poor metabolizers (PMs) of CYP2D6 have a 10-fold higher AUC and a 5-fold higher peak concentration to a given dose of STRATTERA compared with extensive metabolizers (EMs). Approximately 7% of a Caucasian population are PMs. Laboratory tests are available to identify CYP2D6 PMs. The blood levels in PMs are similar to those attained by taking strong inhibitors of CYP2D6. The higher blood levels in PMs lead to a higher rate of some adverse effects of STRATTERA ( see ADVERSE REACTIONS ).
Albuterol --STRATTERA should be administered with caution to patients being treated with systemically-administered (oral or intravenous) albuterol (or other beta 2 agonists) because the action of albuterol on the cardiovascular system can be potentiated.
CYP2D6 inhibitors --Atomoxetine is primarily metabolized by the CYP2D6 pathway to 4-hydroxyatomoxetine. In EMs, selective inhibitors of CYP2D6 increase atomoxetine steady-state plasma concentrations to exposures similar to those observed in PMs. Dosage adjustment of STRATTERA may be necessary when coadministered with CYP2D6 inhibitors, e.g., paroxetine, fluoxetine, and quinidine ( see DOSAGE AND ADMINISTRATION ). In EM individuals treated with paroxetine or fluoxetine, the AUC of atomoxetine is approximately 6- to 8-fold and C ss,max is about 3- to 4-fold greater than atomoxetine alone.
In vitro studies suggest that coadministration of cytochrome P450 inhibitors to PMs will not increase the plasma concentrations of atomoxetine.
Monoamine oxidase inhibitors -- See CONTRAINDICATIONS .
Pressor agents --Because of possible effects on blood pressure, STRATTERA should be used cautiously with pressor agents.
Carcinogenesis --Atomoxetine HCl was not carcinogenic in rats and mice when given in the diet for 2 years at time-weighted average doses up to 47 and 458 mg/kg/day, respectively. The highest dose used in rats is approximately 8 and 5 times the maximum human dose in children and adults, respectively, on a mg/m 2 basis. Plasma levels (AUC) of atomoxetine at this dose in rats are estimated to be 1.8 times (extensive metabolizers) or 0.2 times (poor metabolizers) those in humans receiving the maximum human dose. The highest dose used in mice is approximately 39 and 26 times the maximum human dose in children and adults, respectively, on a mg/m 2 basis.
Mutagenesis --Atomoxetine HCl was negative in a battery of genotoxicity studies that included a reverse point mutation assay (Ames Test), an in vitro mouse lymphoma assay, a chromosomal aberration test in Chinese hamster ovary cells, an unscheduled DNA synthesis test in rat hepatocytes, and an in vivo micronucleus test in mice. However, there was a slight increase in the percentage of Chinese hamster ovary cells with diplochromosomes, suggesting endoreduplication (numerical aberration).
The metabolite N-desmethylatomoxetine HCl was negative in the Ames Test, mouse lymphoma assay, and unscheduled DNA synthesis test.
Impairment of fertility --Atomoxetine HCl did not impair fertility in rats when given in the diet at doses of up to 57 mg/kg/day, which is approximately 6 times the maximum human dose on a mg/m 2 basis.
Pregnancy Category C --Pregnant rabbits were treated with up to 100 mg/kg/day of atomoxetine by gavage throughout the period of organogenesis. At this dose, in 1 of 3 studies, a decrease in live fetuses and an increase in early resorptions was observed. Slight increases in the incidences of atypical origin of carotid artery and absent subclavian artery were observed. These findings were observed at doses that caused slight maternal toxicity. The no-effect dose for these findings was 30 mg/kg/day. The 100-mg/kg dose is approximately 23 times the maximum human dose on a mg/m 2 basis; plasma levels (AUC) of atomoxetine at this dose in rabbits are estimated to be 3.3 times (extensive metabolizers) or 0.4 times (poor metabolizers) those in humans receiving the maximum human dose.
Rats were treated with up to approximately 50 mg/kg/day of atomoxetine (approximately 6 times the maximum human dose on a mg/m 2 basis) in the diet from 2 weeks (females) or 10 weeks (males) prior to mating through the periods of organogenesis and lactation. In 1 of 2 studies, decreases in pup weight and pup survival were observed. The decreased pup survival was also seen at 25 mg/kg (but not at 13 mg/kg). In a study in which rats were treated with atomoxetine in the diet from 2 weeks (females) or 10 weeks (males) prior to mating throughout the period of organogenesis, a decrease in fetal weight (female only) and an increase in the incidence of incomplete ossification of the vertebral arch in fetuses were observed at 40 mg/kg/day (approximately 5 times the maximum human dose on a mg/m 2 basis) but not at 20 mg/kg/day.
No adverse fetal effects were seen when pregnant rats were treated with up to 150 mg/kg/day (approximately 17 times the maximum human dose on a mg/m 2 basis) by gavage throughout the period of organogenesis.
No adequate and well-controlled studies have been conducted in pregnant women. STRATTERA should not be used during pregnancy unless the potential benefit justifies the potential risk to the fetus.
Parturition in rats was not affected by atomoxetine. The effect of STRATTERA on labor and delivery in humans is unknown.
Atomoxetine and/or its metabolites were excreted in the milk of rats. It is not known if atomoxetine is excreted in human milk. Caution should be exercised if STRATTERA is administered to a nursing woman.
The safety and efficacy of STRATTERA in pediatric patients less than 6 years of age have not been established. The efficacy of STRATTERA beyond 9 weeks and safety of STRATTERA beyond 1 year of treatment have not been systematically evaluated.
A study was conducted in young rats to evaluate the effects of atomoxetine on growth and neurobehavioral and sexual development. Rats were treated with 1, 10, or 50 mg/kg/day (approximately 0.2, 2, an 8 times, respectively, the maximum human dose on a mg/m 2 basis) of atomoxetine given by gavage from the early postnatal period (Day 10 of age) through adulthood. Slight delays in onset of vaginal patency (all doses) and preputial separation (10 and 50 mg/kg), slight decreases in epididymal weight and sperm number (10 and 50 mg/kg), and a slight decrease in corpora lutea (50 mg/kg) were seen, but there were no effects on fertility or reproductive performance. A slight delay in onset of incisor eruption was seen at 50 mg/kg. A slight increase in motor activity was seen on Day 15 (males at 10 and 50 mg/kg and females at 50 mg/kg) and on Day 30 (females at 50 mg/kg) but not on Day 60 of age. There were no effects on learning and memory tests. The significance of these findings to humans is unknown.
The safety and efficacy of STRATTERA in geriatric patients have not been established.
STRATTERA was administered to 2067 children or adolescent patients with ADHD and 270 adults with ADHD in clinical studies. During the ADHD clinical trials, 169 patients were treated for longer than 1 year and 526 patients were treated for over 6 months.
The data in the following tables and text cannot be used to predict the incidence of side effects in the course of usual medical practice where patient characteristics and other factors differ from those that prevailed in the clinical trials. Similarly, the cited frequencies cannot be compared with data obtained from other clinical investigations involving different treatments, uses, or investigators. The cited data provide the prescribing physician with some basis for estimating the relative contribution of drug and non-drug factors to the adverse event incidence in the population studied.
Reasons for discontinuation of treatment due to adverse events in child and adolescent clinical trials --In acute child and adolescent placebo-controlled trials, 3.5% (15/427) of atomoxetine subjects and 1.4% (4/294) placebo subjects discontinued for adverse events. For all studies, (including open-label and long-term studies), 5% of extensive metabolizer (EM) patients and 7% of poor metabolizer (PM) patients discontinued because of an adverse event. Among STRATTERA-treated patients, aggression (0.5%, N=2); irritability (0.5%, N=2); somnolence (0.5%, N=2); and vomiting (0.5%, N=2) were the reasons for discontinuation reported by more than 1 patient.
Commonly observed adverse events in acute child and adolescent, placebo-controlled trials --Commonly observed adverse events associated with the use of STRATTERA (incidence of 2% or greater) and not observed at an equivalent incidence among placebo-treated patients (STRATTERA incidence greater than placebo) are listed in Table 1 for the BID trials. Results were similar in the QD trial except as shown in Table 2, which shows both BID and QD results for selected adverse events. The most commonly observed adverse events in patients treated with STRATTERA (incidence of 5% or greater and at least twice the incidence in placebo patients, for either BID or QD dosing) were: dyspepsia, nausea, vomiting, fatigue, appetite decreased, dizziness, and mood swings ( see Tables 1 and 2).
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
The following adverse events occurred in at least 2% of PM patients and were either twice as frequent or statistically significantly more frequent in PM patients compared with EM patients: decreased appetite (23% of PMs, 16% of EMs); insomnia (13% of PMs, 7% of EMs); sedation (4% of PMs, 2% of EMs); depression (6% of PMs, 2% of EMs); tremor (4% of PMs, 1% of EMs); early morning awakening (3% of PMs, 1% of EMs); pruritus (2% of PMs, 1% of EMs); mydriasis (2% of PMs, 1% of EMs).
Reasons for discontinuation of treatment due to adverse events in acute adult placebo-controlled trials --In the acute adult placebo-controlled trials, 8.5% (23/270) atomoxetine subjects and 3.4% (9/266) placebo subjects discontinued for adverse events. Among STRATTERA-treated patients, insomnia (1.1%, N=3); chest pain (0.7%, N=2); palpitations (0.7%, N=2); and urinary retention (0.7%, N=2) were the reasons for discontinuation reported by more than 1 patient.
Commonly observed adverse events in acute adult placebo-controlled trials --Commonly observed adverse events associated with the use of STRATTERA (incidence of 2% or greater) and not observed at an equivalent incidence among placebo-treated patients (STRATTERA incidence greater than placebo) are listed in Table 3. The most commonly observed adverse events in patients treated with STRATTERA (incidence of 5% or greater and at least twice the incidence in placebo patients) were: constipation, dry mouth, nausea, appetite decreased, dizziness, insomnia, decreased libido, ejaculatory problems, impotence, urinary hesitation and/or urinary retention and/or difficulty in micturition, and dysmenorrhea ( see Table 3).
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Male and female sexual dysfunction --Atomoxetine appears to impair sexual function in some patients. Changes in sexual desire, sexual performance, and sexual satisfaction are not well assessed in most clinical trials because they need special attention and because patients and physicians may be reluctant to discuss them. Accordingly, estimates of the incidence of untoward sexual experience and performance cited in product labeling are likely to underestimate the actual incidence. The table below displays the incidence of sexual side effects reported by at least 2% of adult patients taking STRATTERA in placebo-controlled trials.
| |||||||||||||||
There are no adequate and well-controlled studies examining sexual dysfunction with STRATTERA treatment. While it is difficult to know the precise risk of sexual dysfunction associated with the use of STRATTERA, physicians should routinely inquire about such possible side effects.
STRATTERA is not a controlled substance.
In a randomized, double-blind, placebo-controlled, abuse-potential study in adults comparing effects of STRATTERA and placebo, STRATTERA was not associated with a pattern of response that suggested stimulant or euphoriant properties.
Clinical study data in over 2000 children, adolescents, and adults with ADHD and over 1200 adults with depression showed only isolated incidents of drug diversion or inappropriate self-administration associated with STRATTERA. There was no evidence of symptom rebound or adverse events suggesting a drug-discontinuation or withdrawal syndrome.
Drug discrimination studies in rats and monkeys showed inconsistent stimulus generalization between atomoxetine and cocaine.
The effects of overdose greater than twice the maximum recommended daily dose in humans are unknown.
No specific information is available on the treatment of overdose with atomoxetine. Patients who overdose with atomoxetine should be monitored carefully and receive supportive care. Gastric emptying and repeated activated charcoal (with/without cathartics) may prevent systemic absorption.
Dosing of children and adolescents up to 70 kg body weight --STRATTERA should be initiated at a total daily dose of approximately 0.5 mg/kg and increased after a minimum of 3 days to a target total daily dose of approximately 1.2 mg/kg administered either as a single daily dose in the morning or as evenly divided doses in the morning and late afternoon/early evening. No additional benefit has been demonstrated for doses higher than 1.2 mg/kg/day ( see CLINICAL STUDIES ).
The total daily dose in children and adolescents should not exceed 1.4 mg/kg/day or 100 mg, whichever is less.
Dosing of children and adolescents over 70 kg body weight and adults --STRATTERA should be initiated at a total daily dose of 40 mg and increased after a minimum of 3 days to a target total daily dose of approximately 80 mg administered either as a single daily dose in the morning or as evenly divided doses in the morning and late afternoon/early evening. After 2 to 4 additional weeks, the dose may be increased to a maximum of 100 mg in patients who have not achieved an optimal response. There are no data that support increased effectiveness at higher doses ( see CLINICAL STUDIES ).
The maximum recommended total daily dose in children and adolescents over 70 kg and adults is 100 mg.
There is no evidence available from controlled trials to indicate how long the patient with ADHD should be treated with STRATTERA. It is generally agreed, however, that pharmacological treatment of ADHD may be needed for extended periods. Nevertheless, the physician who elects to use STRATTERA for extended periods should periodically reevaluate the long-term usefulness of the drug for the individual patient.
STRATTERA may be taken with or without food.
The safety of single doses over 120 mg and total daily doses above 150 mg have not been systematically evaluated.
Dosing adjustment for hepatically impaired patients --For those ADHD patients who have hepatic insufficiency (HI), dosage adjustment is recommended as follows: For patients with moderate HI (Child-Pugh Class B), initial and target doses should be reduced to 50% of the normal dose (for patients without HI). For patients with severe HI (Child-Pugh Class C), initial dose and target doses should be reduced to 25% of normal ( see Special Populations under CLINICAL PHARMACOLOGY ).
Dosing adjustment for use with a strong CYP2D6 inhibitor --In children and adolescents up to 70 kg body weight administered strong CYP2D6 inhibitors, e.g., paroxetine, fluoxetine, and quinidine, STRATTERA should be initiated at 0.5 mg/kg/day and only increased to the usual target dose of 1.2 mg/kg/day if symptoms fail to improve after 4 weeks and the initial dose is well tolerated.
In children and adolescents over 70 kg body weight and adults administered strong CYP2D6 inhibitors, e.g., paroxetine, fluoxetine, and quinidine, STRATTERA should be initiated at 40 mg/day and only increased to the usual target dose of 80 mg/day if symptoms fail to improve after 4 weeks and the initial dose is well tolerated.
Atomoxetine can be discontinued without being tapered.
STRATTERA capsules are supplied in 10-, 18-, 25-, 40-, and 60-mg strengths.
[see USP Controlled Room Temperature].
| ||||||||||||||||||||||||||||||||||||
Store at 25°C (77°F); excursions permitted to 15° to 30°C (59° to 86°F)
Literature Revised March 5, 2003
PV 3752 AMP
 |
INFORMATION FOR PATIENTS OR THEIR PARENTS OR CAREGIVERS
STRATTERA™
(atomoxetine HCl)
Read this information before you start taking STRATTERA (Stra-TAIR-a). Read this information you get each time you get more STRATTERA. There may be new information. This information does not take the place of talking to your doctor about your medical condition or treatment.
STRATTERA is a non-stimulant medicine used to treat Attention-Deficit/Hyperactivity Disorder (ADHD). STRATTERA contains atomoxetine hydrochloride, a selective norepinephrine reuptake inhibitor. Your doctor has prescribed this medicine as part of an overall treatment plan to control your symptoms of ADHD.
ADHD has 3 main types of symptoms: inattention, hyperactivity, and impulsiveness. Symptoms of inattention include not paying attention, making careless mistakes, not listening, not finishing tasks, not following directions, and being easily distracted. Symptoms of hyperactivity and impulsiveness include fidgeting, talking excessively, running around at inappropriate times, and interrupting others. Some patients have more symptoms of hyperactivity and impulsiveness while others have more symptoms of inattentiveness. Some patients have all 3 types of symptoms.
Symptoms of ADHD in adults may include a lack of organization, problems starting tasks, impulsive actions, daydreaming, daytime drowsiness, slow processing of information, difficulty learning new things, irritability, lack of motivation, sensitivity to criticism, forgetfulness, low self-esteem, and excessive effort to maintain some organization. The symptoms shown by adults who primarily have attention problems but not hyperactivity have been commonly described as Attention-Deficit Disorder (ADD).
Many people have symptoms like these from time to time, but patients with ADHD have these symptoms more than others their age. Symptoms must be present for at least 6 months to be certain of the diagnosis.
Do not take STRATTERA if:
Talk to your doctor before taking STRATTERA if you:
Tell your doctor about all the medicines you take or plan to take, including prescription and non-prescription medicines, dietary supplements, and herbal remedies. Your doctor will decide if you can take STRATTERA with your other medicines.
Certain medicines may change the way your body reacts to STRATTERA. These include medicines used to treat depression [like Paxil ® (paroxetine) and Prozac ® (fluoxetine)], and certain other medicines (like quinidine). Your doctor may need to change your dose of STRATTERA if you are taking it with these medicines.
STRATTERA may change the way your body reacts to oral or intravenous albuterol (or drugs with similar actions), but the effectiveness of these drugs will not be changed. Talk with your doctor before taking STRATTERA if you are taking albuterol.
Call your doctor right away if you take more than your prescribed dose of STRATTERA.
Other important safety information about STRATTERA
Use caution when driving a car or operating heavy machinery until you know how STRATTERA affects you.
Talk to your doctor if you are:
The most common side effects of STRATTERA used in teenagers and children over 6 years old are:
Weight loss may occur after starting STRATTERA. It is not known if growth will be slowed in children who use STRATTERA for a long period of time. Your doctor will watch your weight and height. If you are not growing or gaining weight as expected, your doctor may change your treatment of STRATTERA.
The most common side effects of STRATTERA used in adults are:
Stop taking STRATTERA and call your doctor right away if you get swelling or hives. STRATTERA can cause a serious allergic reaction in rare cases.
This is not a complete list of side effects. Talk to your doctor if you develop any symptoms that concern you.
General advice about STRATTERA
STRATTERA has not been studied in children under 6 years old.
Medicines are sometimes prescribed for conditions that are not mentioned in patient information leaflets. Do not use STRATTERA for a condition for which it was not prescribed. Do not give STRATTERA to other people, even if they have the same symptoms you have.
This leaflet summarizes the most important information about STRATTERA. If you would like more information, talk with your doctor. You can ask your doctor or pharmacist for information on STRATTERA that is written for health professionals. You can also call 1-800-Lilly-Rx (1-800-545-5979) or visit our website at www.strattera.com.
Active ingredient: atomoxetine.
Inactive ingredients: pregelatinized starch, dimethicone, gelatin, sodium lauryl sulfate, FD&C Blue No. 2, synthetic yellow iron oxide, titanium dioxide, and edible black ink.
Store STRATTERA at room temperature.
This patient information summary has been approved by the US Food and Drug Administration.
Literature issued January 17, 2003
www.strattera.com
PV 3740 AMP